
Heterogeneidad tumoral
La heterogeneidad tumoral describe la observación de que diferentes células tumorales pueden mostrar distintos perfiles morfológicos y fenotípicos, que incluyen morfología celular, expresión génica, metabolismo, motilidad, proliferación y potencial metastásico.[1] Este fenómeno ocurre tanto entre tumores (heterogeneidad intertumoral) como dentro de los tumores (heterogeneidad intratumoral). Un nivel mínimo de heterogeneidad intratumoral es una simple consecuencia de la imperfección de la replicación del ADN: siempre que una célula (normal o cancerosa) se divide, se adquieren algunas mutaciones[2] conduce a una población diversa de células cancerosas.[3] La heterogeneidad de las células cancerosas presenta importantes desafíos en el diseño de estrategias de tratamiento eficaces. Sin embargo, la investigación para comprender y caracterizar la heterogeneidad puede permitir una mejor comprensión de las causas y la progresión de la enfermedad. A su vez, esto tiene el potencial de guiar la creación de estrategias de tratamiento más refinadas que incorporen el conocimiento de la heterogeneidad para producir una mayor eficacia.[4]
Se ha observado heterogeneidad tumoral en leucemias,[5] mama,[6] próstata,[7][8][9] colon,[10][11][12] cerebro,[13] esófago,[14] cabeza y cuello,[15] vejiga[16] y carcinomas ginecológicos,[17] liposarcoma,[18] y mieloma múltiple.[19]
Modelos de heterogeneidad
[editar]Se utilizan dos modelos para explicar la heterogeneidad de las células tumorales. Estos son el modelo de células madre cancerosas y el modelo de evolución clonal. Los modelos no son mutuamente excluyentes y se cree que ambos contribuyen a la heterogeneidad en cantidades variables en los diferentes tipos de tumores.[20]
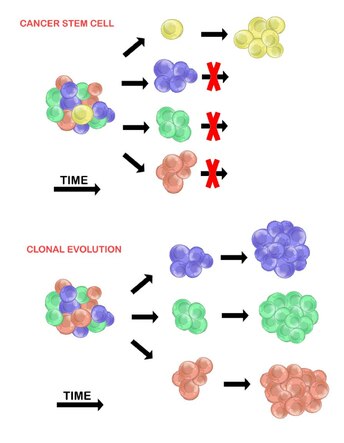
Células madre cancerosas
[editar]El modelo de células madre cancerosas afirma que dentro de una población de células tumorales, solo hay un pequeño subconjunto de células que son tumorigénicas (capaces de formar tumores). Estas células se denominan células madre cancerosas (CSC o cancer stem cells) y están marcadas por la capacidad tanto de autorrenovarse como de diferenciarse en una progenie no tumorigénica. El modelo CSC postula que la heterogeneidad observada entre las células tumorales es el resultado de diferencias en las células madre de las que se originaron. La variabilidad de las células madre a menudo es causada por cambios epigenéticos, pero también puede resultar de la evolución clonal de la población de CSC donde pueden acumularse mutaciones genéticas ventajosas en las CSC y su progenie.[20]
Se ha demostrado evidencia del modelo de células madre cancerosas en múltiples tipos de tumores, como leucemias,[21][22] glioblastoma,[23] cáncer de mama,[24] y cáncer de próstata.[25]
Sin embargo, la existencia de CSC todavía está en debate. Una razón de esto es que los marcadores de CSC han sido difíciles de reproducir en múltiples tumores. Además, los métodos para determinar el potencial tumorigénico utilizan modelos de xenoinjerto. Estos métodos adolecen de limitaciones inherentes tales como la necesidad de controlar la respuesta inmunitaria en el animal trasplantado y la diferencia significativa en las condiciones ambientales desde el sitio del tumor primario hasta el sitio del xenoinjerto (por ejemplo, ausencia de las moléculas exógenas o cofactores requeridos).[26] Esto ha provocado algunas dudas sobre la precisión de los resultados de CSC y las conclusiones sobre qué células tienen potencial tumorigénico.
Evolución clonal
[editar]El modelo de evolución clonal fue propuesto por primera vez en 1976 por Peter Nowell.[27] En este modelo, los tumores surgen de una sola célula mutada, acumulando mutaciones adicionales a medida que avanza. Estos cambios dan lugar a subpoblaciones adicionales, y cada una de estas subpoblaciones tiene la capacidad de dividirse y mutar aún más. Esta heterogeneidad puede dar lugar a subclones que posean una ventaja evolutiva sobre los demás dentro del entorno del tumor, y estos subclones pueden llegar a ser dominantes en el tumor con el tiempo.[28][29] Cuando se propuso, este modelo permitió comprender el crecimiento tumoral, el fracaso del tratamiento y la agresión tumoral que se produce durante el proceso natural de formación del tumor.
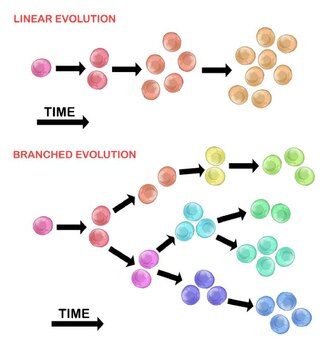
La evolución de la célula tumoral inicial puede ocurrir por dos métodos:
Expansión lineal
[editar]Las mutaciones ordenadas secuencialmente se acumulan en genes impulsores, genes supresores de tumores y enzimas reparadoras del ADN, lo que resulta en la expansión clonal de las células tumorales. Es menos probable que la expansión lineal refleje el criterio de valoración de un tumor maligno[30] porque la acumulación de mutaciones es estocástica en los tumores heterogéneos.
Expansión ramificada
[editar]La expansión a múltiples poblaciones subclonales se produce a través de un mecanismo de división.[28] Este método está más asociado con la heterogeneidad tumoral que con la expansión lineal. La adquisición de mutaciones es aleatoria como resultado de una mayor inestabilidad genómica con cada generación sucesiva. La acumulación mutacional a largo plazo puede proporcionar una ventaja selectiva durante ciertas etapas de la progresión del tumor. El microambiente del tumor también puede contribuir a la expansión del tumor, ya que es capaz de alterar las presiones selectivas a las que están expuestas las células tumorales.[30]
Tipos y causas de heterogeneidad
[editar]Se han observado múltiples tipos de heterogeneidad entre las células tumorales, derivados de la variabilidad tanto genética como no genética.[31]
Heterogeneidad genética
[editar]La heterogeneidad genética es una característica común de los genomas tumorales y puede surgir de múltiples fuentes. Algunos cánceres se inician cuando factores exógenos introducen mutaciones, como la radiación ultravioleta (cánceres de piel) y el tabaco (cáncer de pulmón). Una fuente más común es la inestabilidad genómica, que a menudo surge cuando las vías reguladoras clave se interrumpen en las células. Algunos ejemplos incluyen mecanismos de reparación de ADN alterados que pueden conducir a un aumento de errores de replicación y defectos en la maquinaria de la mitosis que permiten la ganancia o pérdida a gran escala de cromosomas completos.[32] Además, es posible que la variabilidad genética aumente aún más con algunas terapias contra el cáncer (tratamiento con temozolomida y otros fármacos de quimioterapia).[33][34]
La heterogeneidad tumoral mutacional se refiere a variaciones en la frecuencia de mutación en diferentes genes y muestras y puede ser explorada por MutSig Archivado el 3 de octubre de 2017 en Wayback Machine.. La etiología de los procesos mutacionales puede variar considerablemente entre muestras tumorales del mismo o diferentes tipos de cáncer y puede manifestarse en diferentes perfiles mutacionales dependientes del contexto. Puede ser explorado por firmas mutacionales COSMIC o MutaGene.
Otra heterogeneidad
[editar]Las células tumorales también pueden mostrar heterogeneidad entre sus perfiles de expresión. Esto a menudo es causado por cambios epigenéticos subyacentes.[31] Se han detectado variaciones en las firmas de expresión en diferentes regiones de muestras tumorales dentro de un individuo. Los investigadores han demostrado que las mutaciones que afectan convergentes H3K36 metiltransferasa SETD2 y la histona H3K4 desmetilasa KDM5C surgió en secciones tumorales espacialmente separadas. De manera similar, MTOR, un gen que codifica una quinasa reguladora celular, ha demostrado ser constitutivamente activo, aumentando así la fosforilación de S6. Esta fosforilación activa puede servir como biomarcador en el carcinoma de células claras.[30]
La heterogeneidad mecanoquímica es un sello distintivo de las células eucariotas vivas. Tiene un impacto en la regulación de genes epigenéticos. Los procesos mecanoquímicos dinámicos heterogéneos regulan las interrelaciones dentro del grupo de superficies celulares a través de la adhesión.[35] El desarrollo y la propagación del tumor se acompaña de un cambio en la dinámica caótica heterogénea del proceso de interacción mecanoquímica en las células del grupo, incluidas las células dentro del tumor, y es jerárquico para el anfitrión de pacientes con cáncer.[36] Los fenómenos biológicos de heterogeneidad mecanoquímica pueden utilizarse para el diagnóstico diferencial del cáncer gástrico contra pacientes con inflamación de la mucosa gástrica[37] y para aumentar la actividad antimetastásica de las células dendríticas basadas en vacunas cuando se utilizan micropartículas de células tumorales heterogeneizadas mecánicamente para su carga.[38] Existe también un posible abordaje metódico basado en las técnicas diagnósticas y terapéuticas por imagen ecográfica simultánea, respecto al efecto mecanoquímico sobre conglomerados de nanoburbujas con fármacos en el tumor.
Microambiente tumoral
[editar]La heterogeneidad entre las células tumorales puede aumentar aún más debido a la heterogeneidad en el microambiente del tumor. Las diferencias regionales en el tumor (por ejemplo, la disponibilidad de oxígeno) imponen diferentes presiones selectivas sobre las células tumorales, lo que conduce a un espectro más amplio de subclones dominantes en diferentes regiones espaciales del tumor. La influencia del microambiente en la dominancia clonal también es una razón probable de la heterogeneidad entre los tumores primarios y metastásicos observada en muchos pacientes, así como la heterogeneidad inter tumoral observada entre pacientes con el mismo tipo de tumor.[39]
Implicaciones y desafíos
[editar]Resistencia al tratamiento
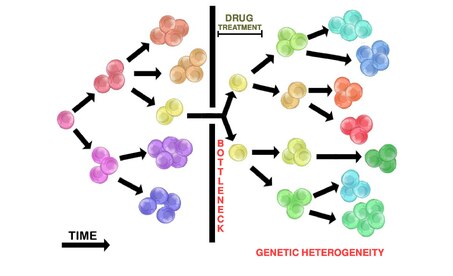
[editar]Los tumores heterogéneos pueden exhibir diferentes sensibilidades a los fármacos citotóxicos entre diferentes poblaciones clonales. Esto se atribuye a interacciones clonales que pueden inhibir o alterar la eficacia terapéutica, lo que plantea un desafío para terapias exitosas en tumores heterogéneos (y sus metástasis heterogéneas).[1]
La administración de fármacos en tumores heterogéneos rara vez destruirá todas las células tumorales. La población tumoral heterogénea inicial puede producir un cuello de botella, de modo que sobrevivirán pocas células resistentes a los fármacos (si las hay). Esto permite que las poblaciones de tumores resistentes se repliquen y desarrollen un nuevo tumor a través del mecanismo de evolución ramificada (ver más arriba). El tumor repoblado resultante es heterogéneo y resistente al tratamiento farmacológico inicial utilizado. El tumor repoblado también puede regresar de una manera más agresiva.
La administración de fármacos citotóxicos a menudo da como resultado una reducción inicial del tumor. Esto representa la destrucción de poblaciones subclonales iniciales no resistentes dentro de un tumor heterogéneo, dejando solo clones resistentes. Estos clones resistentes ahora contienen una ventaja selectiva y pueden replicarse para repoblar el tumor. La replicación probablemente ocurrirá a través de la evolución ramificada, lo que contribuirá a la heterogeneidad del tumor. El tumor repoblado puede parecer más agresivo. Esto se atribuye a la ventaja selectiva de las células tumorales resistentes a los fármacos.
Descubrimiento de biomarcadores
[editar]Debido a las diferencias genéticas dentro y entre los tumores, los biomarcadores que pueden predecir la respuesta al tratamiento o el pronóstico pueden no ser de aplicación generalizada. Sin embargo, se ha sugerido que el nivel de heterogeneidad en sí mismo puede usarse como un biomarcador, ya que es más probable que los tumores más heterogéneos contengan subclones resistentes al tratamiento.[31] Todavía se están realizando más investigaciones sobre el desarrollo de biomarcadores que expliquen la heterogeneidad.
Sistemas modelo
[editar]Los sistemas de modelos actuales generalmente carecen de la heterogeneidad que se observa en los cánceres humanos.[40] Para estudiar con precisión la heterogeneidad tumoral, debemos desarrollar modelos preclínicos más precisos. Uno de estos modelos, el xenoinjerto de tumor derivado del paciente, ha demostrado una excelente utilidad para preservar la heterogeneidad del tumor al tiempo que permite un estudio detallado de los impulsores de la aptitud clonal.[41] Sin embargo, incluso este modelo no puede captar toda la complejidad del cáncer.
Estrategias actuales
[editar]Si bien el problema de identificar, caracterizar y tratar la heterogeneidad tumoral aún se encuentra bajo investigación activa, se han propuesto algunas estrategias efectivas, que incluyen soluciones tanto experimentales como computacionales.
Experimental
[editar]- Enfoque enfocado: analizar un locus genético específico o un conjunto de loci. Esto puede ocurrir mediante la detección de desequilibrios alélicos (el ADN tumoral se compara con el ADN de la línea germinal), la amplificación de regiones cromosómicas (FISH) y/o la secuenciación de genes específicos. Este método se utiliza para rastrear la evolución de una mutación específica de interés o para confirmar una mutación que los investigadores pueden sospechar en un tumor.[1]
- Ventajas: permite el análisis de genes específicos (genes impulsores, supresores de tumores). El proceso es simple con una interpretación directa de los resultados. La FISH y la inmunofluorescencia permiten centrarse en los subtipos de células tumorales.
- Desventajas: un análisis limitado pasará por alto mutaciones importantes adicionales y patrones de expansión clonal. Los desequilibrios alélicos pueden ser difíciles de verificar utilizando marcadores de microsatélites, por lo que requieren verificación mediante una técnica independiente. La FISH requiere una gran cantidad de células y requiere mucha mano de obra.
- Enfoque de todo el genoma: análisis de todo el genoma en muestras de tumores. Esto se puede hacer mediante cariotipo o hibridación genómica comparativa (CGH) para detectar anomalías cromosómicas. La secuenciación de biopsias tumorales es cada vez más común.
- Ventajas: No se basa en conocimientos previos para identificar variantes. El cariotipo identifica regiones de grandes anomalías cromosómicas. CGH proporciona una cobertura imparcial y permite detectar desequilibrios alélicos a pequeña escala (matrices SNP). La secuenciación identificará cualquier variante que contribuya a la heterogeneidad del tumor.
- Desventajas: es difícil determinar el impacto funcional de las variantes (neutrales o patógenas). Resolución limitada. El cariotipo de células cultivadas puede estar sesgado hacia el crecimiento preferencial de subpoblaciones de células tumorales seleccionadas. Resolución limitada en ambos métodos. El enfoque del genoma completo puede generar grandes cantidades de datos y ser difícil de interpretar.
- Estrategia de muestreo multirregional: generalmente requiere múltiples muestras tumorales posquirúrgicas de regiones separadas de un tumor microdisecado. Es importante evitar la contaminación de células no malignas para garantizar una representación precisa de la expresión génica y la composición genética observada únicamente dentro de las células tumorales. El análisis del ADN tumoral dentro de las regiones espacialmente separadas permite la construcción de un modelo evolutivo tridimensional de heterogeneidad tumoral. El muestreo multirregional se utiliza a menudo en combinación con el enfoque de todo el genoma para establecer este modelo de expansión de heterogeneidad 3D.
- Muestreo longitudinal: a través de la progresión del tumor o la progresión del tratamiento, en algunos casos se ha utilizado la obtención de muestras tumorales en múltiples momentos. Esto se ha sugerido como un método confiable para rastrear la evolución clonal.[34][42][43] Sin embargo, esta técnica resulta desafiante en la práctica porque requiere biopsia invasiva periódica. Una nueva investigación sobre la utilización de ADN tumoral libre de células circulantes en la sangre puede proporcionar una forma no invasiva de identificar biomarcadores durante el tratamiento.[44] El muestreo longitudinal utilizado en combinación con el enfoque de todo el genoma permitirá la identificación de las mutaciones acumuladas de células tumorales a lo largo del tiempo. Esto, a su vez, puede identificar las mutaciones impulsoras clave (observadas en las muestras iniciales de tumores).
- La terapia adaptativa puede usarse para prevenir un mayor crecimiento tumoral ajustando la dosis del fármaco y el momento de administración del fármaco en función de la respuesta del tumor. Se supone que esta estrategia evita que las variantes resistentes dominen un tumor. Sin embargo, se requiere más investigación sobre su aplicabilidad.[45]
Secuenciación
[editar]- Se puede utilizar la secuenciación de tumores a granel, donde el ADN se extrae de una mezcla de células tumorales y se analiza de una vez. La presencia de poblaciones de tumores heterogéneas (subclones) introduce desafíos adicionales como:
- La incapacidad para detectar mutaciones en subclones raros. Dado que estas mutaciones ocurrirán con baja frecuencia en la muestra combinada, pueden ser indistinguibles del ruido de fondo. Sin embargo, se están desarrollando activamente muchas variantes de llamadas que están diseñadas específicamente para datos de cáncer y tienen como objetivo identificar variantes raras presentes en poblaciones subclonales más pequeñas.[46][47][48][49] Por lo general, estos utilizan ADN normal emparejado como un medio para distinguir la variación somática verdadera de la variación de la línea germinal y el error de secuenciación de fondo.
- La incapacidad para determinar qué subclones contienen cada mutación. Dado que los datos se agrupan, no está claro qué mutaciones coexisten y de qué poblaciones se originan. Se están desarrollando nuevas herramientas que intentan resolver la estructura clonal utilizando frecuencias alélicas para las mutaciones observadas.[50]
- La secuenciación unicelular es una técnica nueva que es valiosa para evaluar la heterogeneidad tumoral porque puede caracterizar células tumorales individuales. Esto significa que el perfil mutacional completo de múltiples células distintas se puede determinar sin ambigüedad. Si bien con la tecnología actual, es difícil evaluar un número suficientemente grande de células individuales para obtener poder estadístico, los datos de tumores unicelulares tienen múltiples ventajas, que incluyen:
- La capacidad de construir un árbol filogenético que muestre la evolución de las poblaciones tumorales. Usando secuencias de genoma completo o pseudo-secuencias basadas en SNP de células individuales, se puede estimar la evolución de los subclones. Esto permite la identificación de poblaciones que han persistido a lo largo del tiempo y puede reducir la lista de mutaciones que potencialmente confieren una ventaja de crecimiento o resistencia al tratamiento en subclones específicos.[51] Los algoritmos para inferir una filogenia tumoral a partir de datos de secuenciación de ADN de una sola célula incluyen SCITE,[52] OncoNEM,[53] SiFit,[54] SiCloneFit,[55] PhISCS,[56] y PhISCS-BnB.[57]
- La secuenciación de secciones se puede realizar en múltiples porciones de un solo tumor sólido, y la variación en las frecuencias de mutación entre las secciones se puede analizar para inferir la estructura clonal. Las ventajas de este enfoque sobre la secuenciación única incluyen más poder estadístico y disponibilidad de información más precisa sobre el posicionamiento espacial de las muestras. Este último puede usarse para inferir la frecuencia de clones en secciones y proporcionar información sobre cómo evoluciona un tumor en el espacio. Para inferir los genotipos de los clones y los árboles filogenéticos que modelan la evolución de un tumor en el tiempo, se desarrollaron varios métodos computacionales[58][59][60] incluyendo Clomial,[61] cloneHD,[62] PhyloWGS,[63] PyClone,[64] Cloe,[65] phyC,[66] Canopy,[67] TargetClone, ddClone,[68] PASTRI,[69] GLClone,[70] TRaIT, WSCUnmix,[71] B-SCITE., ThetA,[72] SIFA,[73] Sclust,[74] SeqClone,[75] CALDER,[76] BAMSE,[77] Meltos,[78] SubMARine,[79] y RNDCLONE.[80]
Véase también
[editar]Referencias
[editar]- ↑ a b c Marusyk, A; Polyak, K (2010). «Tumor heterogeneity: Causes and consequences». Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1805 (1): 105-117. PMC 2814927. PMID 19931353. doi:10.1016/j.bbcan.2009.11.002.
- ↑ Vogelstein, Bert; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. (2013). «Cancer Genome Landscapes». Science 373 (6127): 1546-1556. Bibcode:2013Sci...339.1546V. PMC 3749880. PMID 23539594. doi:10.1126/science.1235122.
- ↑ Heppner, G.A. (1984). «Tumor Heterogeneity». Cancer Research 44 (6): 2259-2265. PMID 6372991.
- ↑ Reiter, Johannes G; Makohon-Moore, Alvin P; Gerold, Jeffrey M; Heyde, Alexander; Attiyeh, Marc A; Kohutek, Zachary A; Tokheim, Collin J; Brown, Alexia et al. (2018). «Minimal functional driver gene heterogeneity among untreated metastases». Science 361 (6406): 1033-1037. PMC 6329287. PMID 30190408. doi:10.1126/science.aat7171.
- ↑ Campbell, P. J.; Pleasance, E. D.; Stephens, P. J.; Dicks, E; Rance, R; Goodhead, I; Follows, G. A.; Green, A. R. et al. (2008). «Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing». Proceedings of the National Academy of Sciences 105 (35): 13081-13086. Bibcode:2008PNAS..10513081C. PMC 2529122. PMID 18723673. doi:10.1073/pnas.0801523105.
- ↑ Shipitsin, M; Campbell, L. L.; Argani, P; Weremowicz, S; Bloushtain-Qimron, N; Yao, J; Nikolskaya, T; Serebryiskaya, T et al. (2007). «Molecular definition of breast tumor heterogeneity». Cancer Cell 11 (3): 259-273. PMID 17349583. doi:10.1016/j.ccr.2007.01.013.
- ↑ MacIntosh, C. A.; Stower, M; Reid, N; Maitland, N. J. (1998). «Precise microdissection of human prostate cancers reveals genotypic heterogeneity». Cancer Research 58 (1): 23-28. PMID 9426051.
- ↑ Alvarado, C; Beitel, L. K.; Sircar, K; Aprikian, A; Trifiro, M; Gottlieb, B (2005). «Somatic mosaicism and cancer: A micro-genetic examination into the role of the androgen receptor gene in prostate cancer». Cancer Research 65 (18): 8514-8518. PMID 16166332. doi:10.1158/0008-5472.CAN-05-0399.
- ↑ Konishi, N; Hiasa, Y; Matsuda, H; Tao, M; Tsuzuki, T; Hayashi, I; Kitahori, Y; Shiraishi, T et al. (1995). «Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma». The American Journal of Pathology 147 (4): 1112-1122. PMC 1871010. PMID 7573356.
- ↑ González-García, I; Solé, R. V.; Costa, J (2002). «Metapopulation dynamics and spatial heterogeneity in cancer». Proceedings of the National Academy of Sciences 99 (20): 13085-13089. Bibcode:2002PNAS...9913085G. PMC 130590. PMID 12351679. doi:10.1073/pnas.202139299.
- ↑ Samowitz, W. S.; Slattery, M. L. (1999). «Regional reproducibility of microsatellite instability in sporadic colorectal cancer». Genes, Chromosomes and Cancer 26 (2): 106-114. PMID 10469448. doi:10.1002/(SICI)1098-2264(199910)26:2<106::AID-GCC2>3.0.CO;2-F.
- ↑ Giaretti, W; Monaco, R; Pujic, N; Rapallo, A; Nigro, S; Geido, E (1996). «Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: Association with degree of DNA aneuploidy». The American Journal of Pathology 149 (1): 237-245. PMC 1865212. PMID 8686748.
- ↑ Heppner, G. H. (1984). «Tumor heterogeneity». Cancer Research 44 (6): 2259-2265. PMID 6372991.
- ↑ Maley, C. C.; Galipeau, P. C.; Finley, J. C.; Wongsurawat, V. J.; Li, X; Sanchez, C. A.; Paulson, T. G.; Blount, P. L. et al. (2006). «Genetic clonal diversity predicts progression to esophageal adenocarcinoma». Nature Genetics 38 (4): 468-473. PMID 16565718. doi:10.1038/ng1768.
- ↑ Califano, J; Van Der Riet, P; Westra, W; Nawroz, H; Clayman, G; Piantadosi, S; Corio, R; Lee, D et al. (1996). «Genetic progression model for head and neck cancer: Implications for field cancerization». Cancer Research 56 (11): 2488-2492. PMID 8653682.
- ↑ Sauter, G; Moch, H; Gasser, T. C.; Mihatsch, M. J.; Waldman, F. M. (1995). «Heterogeneity of chromosome 17 and erbB-2 gene copy number in primary and metastatic bladder cancer». Cytometry 21 (1): 40-46. PMID 8529469. doi:10.1002/cyto.990210109.
- ↑ Fujii, H; Yoshida, M; Gong, Z. X.; Matsumoto, T; Hamano, Y; Fukunaga, M; Hruban, R. H.; Gabrielson, E et al. (2000). «Frequent genetic heterogeneity in the clonal evolution of gynecological carcinosarcoma and its influence on phenotypic diversity». Cancer Research 60 (1): 114-120. PMID 10646862.
- ↑ Horvai, A. E.; Devries, S; Roy, R; O'Donnell, R. J.; Waldman, F (2009). «Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma». Modern Pathology 22 (11): 1477-1488. PMID 19734852. doi:10.1038/modpathol.2009.119.
- ↑ Pantou, D; Rizou, H; Tsarouha, H; Pouli, A; Papanastasiou, K; Stamatellou, M; Trangas, T; Pandis, N et al. (2005). «Cytogenetic manifestations of multiple myeloma heterogeneity». Genes, Chromosomes and Cancer 42 (1): 44-57. PMID 15495197. doi:10.1002/gcc.20114.
- ↑ a b Shackleton, M; Quintana, E; Fearon, E. R.; Morrison, S. J. (2009). «Heterogeneity in cancer: Cancer stem cells versus clonal evolution». Cell 138 (5): 822-829. PMID 19737509. doi:10.1016/j.cell.2009.08.017.
- ↑ Lapidot, T; Sirard, C; Vormoor, J; Murdoch, B; Hoang, T; Caceres-Cortes, J; Minden, M; Paterson, B et al. (1994). «A cell initiating human acute myeloid leukaemia after transplantation into SCID mice». Nature 367 (6464): 645-648. Bibcode:1994Natur.367..645L. PMID 7509044. doi:10.1038/367645a0.
- ↑ Wang, J. C.; Lapidot, T; Cashman, J. D.; Doedens, M; Addy, L; Sutherland, D. R.; Nayar, R; Laraya, P et al. (1998). «High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase». Blood 91 (7): 2406-2414. PMID 9516140. doi:10.1182/blood.V91.7.2406.
- ↑ Singh, S. K.; Hawkins, C; Clarke, I. D.; Squire, J. A.; Bayani, J; Hide, T; Henkelman, R. M.; Cusimano, M. D. et al. (2004). «Identification of human brain tumour initiating cells». Nature 432 (7015): 396-401. Bibcode:2004Natur.432..396S. PMID 15549107. doi:10.1038/nature03128.
- ↑ Al-Hajj, M; Wicha, M. S.; Benito-Hernandez, A; Morrison, S. J.; Clarke, M. F. (2003). «Prospective identification of tumorigenic breast cancer cells». Proceedings of the National Academy of Sciences 100 (7): 3983-3988. Bibcode:2003PNAS..100.3983A. PMC 153034. PMID 12629218. doi:10.1073/pnas.0530291100.
- ↑ Maitland, N. J.; Collins, A. T. (2008). «Prostate cancer stem cells: A new target for therapy». Journal of Clinical Oncology 26 (17): 2862-2870. PMID 18539965. doi:10.1200/JCO.2007.15.1472.
- ↑ Meacham, C. E.; Morrison, S. J. (2013). «Tumour heterogeneity and cancer cell plasticity». Nature 501 (7467): 328-337. Bibcode:2013Natur.501..328M. PMC 4521623. PMID 24048065. doi:10.1038/nature12624.
- ↑ Nowell, P. C. (1976). «The clonal evolution of tumor cell populations». Science 194 (4260): 23-28. Bibcode:1976Sci...194...23N. PMID 959840. doi:10.1126/science.959840.
- ↑ a b Swanton, C (2012). «Intratumor heterogeneity: Evolution through space and time». Cancer Research 72 (19): 4875-4882. PMC 3712191. PMID 23002210. doi:10.1158/0008-5472.CAN-12-2217.
- ↑ Merlo, L. M. F.; Pepper, J. W.; Reid, B. J.; Maley, C. C. (2006). «Cancer as an evolutionary and ecological process». Nature Reviews Cancer 6 (12): 924-935. PMID 17109012. doi:10.1038/nrc2013.
- ↑ a b c Gerlinger, M; Rowan, A. J.; Horswell, S; Larkin, J; Endesfelder, D; Gronroos, E; Martinez, P; Matthews, N et al. (2012). «Intratumor heterogeneity and branched evolution revealed by multiregion sequencing». New England Journal of Medicine 366 (10): 883-892. PMC 4878653. PMID 22397650. doi:10.1056/NEJMoa1113205.
- ↑ a b c Marusyk, A; Almendro, V; Polyak, K (2012). «Intra-tumour heterogeneity: A looking glass for cancer?». Nature Reviews Cancer 12 (5): 323-334. PMID 22513401. doi:10.1038/nrc3261.
- ↑ Burrell, R. A.; McGranahan, N; Bartek, J; Swanton, C (2013). «The causes and consequences of genetic heterogeneity in cancer evolution». Nature 501 (7467): 338-345. Bibcode:2013Natur.501..338B. PMID 24048066. doi:10.1038/nature12625.
- ↑ Johnson, B. E.; Mazor, T; Hong, C; Barnes, M; Aihara, K; McLean, C. Y.; Fouse, S. D.; Yamamoto, S et al. (2014). «Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma». Science 343 (6167): 189-193. Bibcode:2014Sci...343..189J. PMC 3998672. PMID 24336570. doi:10.1126/science.1239947.
- ↑ a b Ding, L; Ley, T. J.; Larson, D. E.; Miller, C. A.; Koboldt, D. C.; Welch, J. S.; Ritchey, J. K.; Young, M. A. et al. (2012). «Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing». Nature 481 (7382): 506-510. Bibcode:2012Natur.481..506D. PMC 3267864. PMID 22237025. doi:10.1038/nature10738.
- ↑ G.M.Edelman (1989). «Topobiology». J. Scientific American 260 (5): 76-88. PMID 2717916. doi:10.1038/scientificamerican0589-76.
- ↑ V.E. Orel; N.N Dzyatkovskaya; M.I. Danko; A.V. Romanov; Y.I. Mel'nik; Y.A. Grinevich; S.V. Martynenko (2004). «Spatial and mechanoemission chaos of mechanically deformed tumor cells». J. Journal of Mechanics in Medicine and Biology 4 (1): 31-45. doi:10.1142/s0219519404000886.
- ↑ V.E. Orel; A.V. Romanov; N.N. Dzyatkovskaya; Yu.I. Mel’nik (2002). «The device and algorithm for estimation of the mechanoemisson chaos in blood of patients with gastric cancer». J. Medical Engineering & Physics 24 (5): 365-371. PMID 12052364. doi:10.1016/s1350-4533(02)00022-x.
- ↑ N. Khranovskaya; V. Orel; Y. Grinevich; O. Alekseenko; A. Romanov; O. Skachkova; N.Dzyatkovskaya; A. Burlaka et al. (2012). «Mechanical heterogenization of Lewis lung carcinoma cells can improve antimetastatic effect of dendritic cells». J. Journal of Mechanics in Medicine and Biology 3 (12): 22. doi:10.1142/S0219519411004757.
- ↑ Junttila, M. R.; De Sauvage, F. J. (2013). «Influence of tumour micro-environment heterogeneity on therapeutic response». Nature 501 (7467): 346-354. Bibcode:2013Natur.501..346J. PMID 24048067. doi:10.1038/nature12626.
- ↑ Auman, James Todd; McLeod, Howard L. (1 de enero de 2010). «Colorectal Cancer Cell Lines Lack the Molecular Heterogeneity of Clinical Colorectal Tumors». Clinical Colorectal Cancer 9 (1): 40-47. PMID 20100687. doi:10.3816/ccc.2010.n.005.
- ↑ Cassidy, John W.; Caldas, Carlos; Bruna, Alejandra (1 de agosto de 2015). «Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts». Cancer Research 75 (15): 2963-2968. ISSN 0008-5472. PMC 4539570. PMID 26180079. doi:10.1158/0008-5472.CAN-15-0727.
- ↑ «Integrated genomic characterization of IDH1-mutant glioma malignant progression». Nature Genetics 48 (1): 59-66. Nov 2015. PMC 4829945. PMID 26618343. doi:10.1038/ng.3457.
- ↑ Bedard, P. L.; Hansen, A. R.; Ratain, M. J.; Siu, L. L. (2013). «Tumour heterogeneity in the clinic». Nature 501 (7467): 355-364. Bibcode:2013Natur.501..355B. PMC 5224525. PMID 24048068. doi:10.1038/nature12627.
- ↑ Dawson, S. J.; Tsui, D. W.; Murtaza, M; Biggs, H; Rueda, O. M.; Chin, S. F.; Dunning, M. J.; Gale, D et al. (2013). «Analysis of circulating tumor DNA to monitor metastatic breast cancer». New England Journal of Medicine 368 (13): 1199-1209. PMID 23484797. doi:10.1056/NEJMoa1213261.
- ↑ Gatenby, R. A.; Silva, A. S.; Gillies, R. J.; Frieden, B. R. (2009). «Adaptive therapy». Cancer Research 69 (11): 4894-4903. PMC 3728826. PMID 19487300. doi:10.1158/0008-5472.CAN-08-3658.
- ↑ Cibulskis, K; Lawrence, M. S.; Carter, S. L.; Sivachenko, A; Jaffe, D; Sougnez, C; Gabriel, S; Meyerson, M et al. (2013). «Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples». Nature Biotechnology 31 (3): 213-219. PMC 3833702. PMID 23396013. doi:10.1038/nbt.2514.
- ↑ Koboldt, D. C.; Zhang, Q; Larson, D. E.; Shen, D; McLellan, M. D.; Lin, L; Miller, C. A.; Mardis, E. R. et al. (2012). «Var Scan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing». Genome Research 22 (3): 568-576. PMC 3290792. PMID 22300766. doi:10.1101/gr.129684.111.
- ↑ Saunders, C. T.; Wong, W. S.; Swamy, S; Becq, J; Murray, L. J.; Cheetham, R. K. (2012). «Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs». Bioinformatics 28 (14): 1811-1817. PMID 22581179. doi:10.1093/bioinformatics/bts271.
- ↑ Carter, S. L.; Cibulskis, K; Helman, E; McKenna, A; Shen, H; Zack, T; Laird, P. W.; Onofrio, R. C. et al. (2012). «Absolute quantification of somatic DNA alterations in human cancer». Nature Biotechnology 30 (5): 413-421. PMC 4383288. PMID 22544022. doi:10.1038/nbt.2203.
- ↑ Shah, S. P.; Roth, A; Goya, R; Oloumi, A; Ha, G; Zhao, Y; Turashvili, G; Ding, J et al. (2012). «The clonal and mutational evolution spectrum of primary triple-negative breast cancers». Nature 486 (7403): 395-399. Bibcode:2012Natur.486..395S. PMC 3863681. PMID 22495314. doi:10.1038/nature10933.
- ↑
- ↑ Jahn, Katharina (2016). «Tree inference for single-cell data». Genome Biology 17: 86. PMC 4858868. PMID 27149953. doi:10.1186/s13059-016-0936-x.
- ↑ Ross, Edith (2016). «OncoNEM: inferring tumor evolution from single-cell sequencing data». Genome Biology 17: 69. PMC 4832472. PMID 27083415. doi:10.1186/s13059-016-0929-9.
- ↑ Zafar, Hamim (2017). «SiFit: inferring tumor trees from single-cell sequencing data under finite-sites models». Genome Biology 18 (1): 178. PMC 5606061. PMID 28927434. doi:10.1186/s13059-017-1311-2.
- ↑ Zafar, Hamim (2019). «SiCloneFit: Bayesian inference of population structure, genotype, and phylogeny of tumor clones from single-cell genome sequencing data». Genome Research 29 (11): 1847-1859. PMC 6836738. PMID 31628257. doi:10.1101/gr.243121.118.
- ↑ Malikic, Salem; Rashidi Mehrabadi, Farid (2019). «PhISCS: a combinatorial approach for subperfect tumor phylogeny reconstruction via integrative use of single-cell and bulk sequencing data». Genome Research 29 (11): 1860-1877. PMID 31628256. doi:10.1101/gr.234435.118.
- ↑ Sadeqi Azer, Erfan; Rashidi Mehrabadi, Farid (2020). «PhISCS-BnB: a fast branch and bound algorithm for the perfect tumor phylogeny reconstruction problem». Bioinformatics 36 (Supplement_1): i169-i176. PMID 32657358. doi:10.1093/bioinformatics/btaa464.
- ↑ Kuipers, Jack (2017). «Advances in understanding tumour evolution through single-cell sequencing». Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1867 (2): 127-138. PMC 5813714. PMID 28193548. doi:10.1016/j.bbcan.2017.02.001.
- ↑ Schwartz, Russell (13 de febrero de 2017). «The evolution of tumour phylogenetics: principles and practice». Nature Reviews Genetics 18 (4): 213-229. PMC 5886015. PMID 28190876. doi:10.1038/nrg.2016.170.
- ↑ Farahani, Hossein; de Souza, Camila P. E.; Billings, Raewyn; Yap, Damian; Shumansky, Karey; Wan, Adrian; Lai, Daniel; Mes-Masson, Anne-Marie et al. (18 de octubre de 2017). «Engineered in-vitro cell line mixtures and robust evaluation of computational methods for clonal decomposition and longitudinal dynamics in cancer». Scientific Reports 7 (1): 13467. Bibcode:2017NatSR...713467F. PMC 5647443. PMID 29044127. doi:10.1038/s41598-017-13338-8.
- ↑ Zare, Habil (2014). «Inferring clonal composition from multiple sections of a breast cancer». PLOS Computational Biology 10 (7): e1003703. Bibcode:2014PLSCB..10E3703Z. PMC 4091710. PMID 25010360. doi:10.1371/journal.pcbi.1003703.
- ↑ Fischer, Andrej (2014). «High-definition reconstruction of clonal composition in cancer». Cell Reports 7 (5): 1740-1752. PMC 4062932. PMID 24882004. doi:10.1016/j.celrep.2014.04.055.
- ↑ Deshwar, Amit (2015). «Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns». Genome Biology 16: 35. PMC 4359439. PMID 25786235. doi:10.1186/s13059-015-0602-8.
- ↑ Roth, Andrew (2014). «PyClone: statistical inference of clonal population structure in cancer». Nature Methods 11 (4): 396-398. PMC 4864026. PMID 24633410. doi:10.1038/nmeth.2883.
- ↑ Marass, Francesco (2015). «A phylogenetic latent feature model for clonal deconvolution». The Annals of Applied Statistics 10 (4): 2377-2404. arXiv:1604.01715. doi:10.1214/16-AOAS986.
- ↑ Matsui, Yusuke (2016). «phyC: Clustering cancer evolutionary trees». PLOS Computational Biology 13 (5): e1005509. Bibcode:2017PLSCB..13E5509M. PMC 5432190. PMID 28459850. doi:10.1371/journal.pcbi.1005509.
- ↑ Jiang, Yuchao; Qiu, Yu; Minn, Andy J.; Zhang, Nancy R. (29 de agosto de 2016). «Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing». Proceedings of the National Academy of Sciences 113 (37): E5528-37. PMC 5027458. PMID 27573852. doi:10.1073/pnas.1522203113.
- ↑ Salehi, Sohrab (2017). «ddClone: joint statistical inference of clonal populations from single cell and bulk tumour sequencing data». Genome Biology 18 (1): 44. PMC 5333399. PMID 28249593. doi:10.1186/s13059-017-1169-3.
- ↑ Satas, Gryte (2017). «Tumor phylogeny inference using tree-constrained importance sampling». Bioinformatics 33 (14): i152-i160. PMC 5870673. PMID 28882002. doi:10.1093/bioinformatics/btx270.
- ↑ Geng, Yu (2017). «Identifying Heterogeneity Patterns of Allelic Imbalance on Germline Variants to Infer Clonal Architecture». International Conference on Intelligent Computing. Lecture Notes in Computer Science 10362: 286-297. ISBN 978-3-319-63311-4. doi:10.1007/978-3-319-63312-1_26.
- ↑ Roman, Theodore; Xie, Lu; Schwartz, Russell; Raphael, Benjamin J. (23 de octubre de 2017). «Automated deconvolution of structured mixtures from heterogeneous tumor genomic data». PLOS Computational Biology 13 (10): e1005815. Bibcode:2017PLSCB..13E5815R. PMC 5695636. PMID 29059177. arXiv:1604.02487. doi:10.1371/journal.pcbi.1005815.
- ↑ Oesper, Layla; Mahmoody, Ahmad; Raphael, Benjamin J. (29 de julio de 2013). «THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data». Genome Biology 14 (7): R80. ISSN 1474-760X. PMC 4054893. PMID 23895164. doi:10.1186/gb-2013-14-7-r80.
- ↑ Zeng, By (2018). «Phylogeny-based tumor subclone identification using a Bayesian feature allocation model». MISSING LINK..
- ↑ Cun, Yupeng; Yang, Tsun-Po; Achter, Viktor; Lang, Ulrich; Peifer, Martin (24 de mayo de 2018). «Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust». Nature Protocols 13 (6): 1488-1501. ISSN 1754-2189. PMID 29844525. doi:10.1038/nprot.2018.033.
- ↑ Wang, Xiaodong; Ogundijo, Oyetunji E. (1 de diciembre de 2019). «SeqClone: sequential Monte Carlo based inference of tumor subclones». BMC Bioinformatics (en inglés) 20 (1): 6. ISSN 1471-2105. PMC 6320595. PMID 30611189. doi:10.1186/s12859-018-2562-y.
- ↑ Raphael, Benjamin J.; Satas, Gryte; Myers, Matthew A. (22 de enero de 2019). «Inferring tumor evolution from longitudinal samples». bioRxiv (en inglés): 526814. doi:10.1101/526814.
- ↑ Toosi, Hosein; Moeini, Ali; Hajirasouliha, Iman (6 de junio de 2019). «BAMSE: Bayesian model selection for tumor phylogeny inference among multiple samples». BMC Bioinformatics 20 (11): 282. ISSN 1471-2105. PMC 6551234. PMID 31167637. doi:10.1186/s12859-019-2824-3.
- ↑ Ricketts, Camir; Seidman, Daniel; Popic, Victoria; Hormozdiari, Fereydoun; Batzoglou, Serafim; Hajirasouliha, Iman (4 de octubre de 2019). «Meltos: Multi-Sample Tumor Phylogeny Reconstruction for Structural Variants». Bioinformatics 36 (4): 1082-1090. PMID 31584621. doi:10.1093/bioinformatics/btz737.
- ↑ Sundermann, Linda. «Reconstructing tumor evolutionary histories and clone trees in polynomial-time with SubMARine». bioRxiv. Consultado el 22 de junio de 2020.
- ↑ Zhou, Tianjian. «RNDCLONE: TUMOR SUBCLONE RECONSTRUCTION BASED ON INTEGRATING DNA AND RNA SEQUENCE DATA». Consultado el 23 de agosto de 2020.
| Control de autoridades |
|
|---|
 Datos: Q15917291
Datos: Q15917291